Nuevas dianas terapéuticas contra el cáncer
noviembre 2, 2007 at 11:55 am Deja un comentario
El progreso que la Oncología Médica ha realizado en los últimos años se ha debido fundamentalmente al campo del desarrollo de fármacos antineoplásicos, especialmente en la esfera de la quimioterapia y la hormonoterapia. Estos fármacos están diseñados para producir una máxima destrucción tumoral, aunque en muchos casos su mecanismo íntimo de acción no se ha conocido sino muy recientemente. Por lo tanto fueron investigados como fármacos antitumorales, en base la mayoría de las veces, a estudios experimentales empíricos y no diseñados ad hoc. Pero es que además en los últimos años se ha podido demostrar que tales fármacos actúan como poderosos anti-apoptóticos o como inductores de p53. Por lo tanto sin saberlo se estaban logrando fármacos que tenían potencial no sólo destructor sino además capaces de alterar el fenotipo y el genotipo tumoral.
Hoy se entiende el cáncer como un conjunto de enfermedades con capacidad ilimitada de proliferarse y con profundos errores en los mecanismos de división, y específicamente en la entrada y salida en el ciclo celular. Es decir que los fenómenos de señalización (factores de crecimiento, receptores, proteínas citoplasmáticas), división celular (ciclinas, genes supresores), control de apoptosis, etc, están íntimamente implicados en el desarrollo del cáncer. Aún más, los tumores una vez establecidos necesitan de una formación de vasos nuevos (neo-angiogénesis) que le asegurará su supervivencia.
En base a todo ello el tratamiento médico del cáncer del futuro debe asentarse sobre unas bases racionales del conocimiento de la proliferación, diferenciación, y apoptosis, y los fármacos deben ser diseñados de entrada de acuerdo a su mecanismo de acción y conociendo al detalle las funciones que pueden llegar a controlar (Figura 1 y Figura 2). Serán por tanto fundamentalmente inhibidores de señales, ya que el diseño de fármacos activadores es mucho más complicado.
|
Figura 1. Los nuevos agentes terapéuticos basados en nuevas dianas moleculares o citostáticos, son capaces, al menos en teoría de transformar las células cancerosas en células diferenciadas no proliferativas pero viables, en células diferenciadas terminales que acaban en apoptosis y en células en estado quiescente o dormido. |

|
Figura 2. Mientras los citotóxicos tienen como fin último la destrucción de la célula tumoral al producir importantes alteraciones en el DNA o en los microtúbulos, los agentes citostáticos actúan sobre las vías de señalización intracelular, la inducción de apoptosis, la alteración de la angiogénesis o por interacción celular. |

A continuación se mencionan las más importantes áreas de desarrollo en el campo del tratamiento de los tumores sólidos:
Receptores de los factores de crecimiento epidérmico
La división y proliferación celular están controladas por vías de señalización que se inician con la unión de los factores de crecimiento con los receptores correspondientes. En la transformación cancerosa, los receptores de los factores de crecimiento de tipo epidérmico juegan un papel de gran relevancia y corresponden a 4 grupos: el ErB-1 o Her1 (EGFR, propiamente dicho); el ErbB-2 (Her2-neu); el ErB-3 (Her3), y el ErbB-4(Her4). Los ligandos de estos receptores son el EGF, TGF-a, HB-EGF, AR, epiregulina, y VGF para el EGFR; las herregulinas para el Her-3; y NRG2, NRG3, herregulinas y B-celulina para el ErbB-4. El Her-2 es un receptor huérfano. Todos los receptores constan de dominios extracitoplasmáticos ricos en cisteína que tras la unión con los ligandos son dimerizados, e incluso heterodimerizados, y que dan lugar a la fosforilización posterior del dominio catalítico intracitoplasmático rico en tirosín quinasa, iniciándose de esta manera todo el proceso de señalización intracelular (Figura 3). Estos receptores se encuentran muy comúnmente sobreexpresados e incluso transactivados. Concretamente el EGFR se encuentra sobreexpresado en el 40-80% de los casos de cáncer de pulmón de célula no pequeña (NSCL), 40-80% de cáncer de próstata, 33-74% de los cánceres gástricos, 14-91% en mama, 25-77% en colon, 30-50% en páncreas y 35-70% en cáncer de ovario. Lo anterior se relaciona con la invasión, metástasis, estadios avanzados de la enfermedad, resistencia a la quimioterapia, y pobre respuesta a la hormonoterapia, etc. Se trata por tanto de un punto crítico de activación que define la posterior proliferación, apoptosis, angiogénesis, y en definitiva la presencia de metástasis (Figura 4). Por lo tanto, no es de extrañar que dentro de las nuevas dianas terapéuticas un gran foco de atención han sido dichos receptores. Teóricamente pueden desarrollarse anticuerpos monoclonales que impidan la unión de los factores de crecimiento con los receptores correspondientes, inhibidores específicos de tirosina quinasas, conjugados de toxinas e incluso
|
Figura 3. |

|
Figura 4.(Modificada de Baselga). La señalización intracelular mediada por los receptores del factor de crecimiento epidérmico (EGFR) implica la participación en cascada de numerosos genes, bien vía RAS y por tanto las MAK kinasas, o a través de PI3-K y AKT. De esa manera se transmite la señal al núcleo donde se produce la transcripción de genes y la progresión en el ciclo celular. En las células cancerosas los EGFR pueden estar mutados y por tanto activados constitutivamente. De esa manera la señal es permanente y se induce proliferación, resistencia a quimioterapia y radioterapia, fenómenos anti-apoptóticos, angiogénesis y los fenómenos de invasión y metástasis. |

|
Figura 5. Las posibilidades terapéuticas orientadas a bloquear la señal vía los EGFR se puede realizar con anticuerpos monoclonales, inhibidores de tirosina quinasas y oligonucleótidos antisentido. |

El éxito de los anticuerpos monoclonales ya ha quedado demostrado con el desarrollo del Trastuzumab, un anticuerpo monoclonal dirigido frente a una proteína de 185 KdA que actúa como receptor transmembrana de tirosina quinasa, el receptor Her-2 (c-erbB-2 neu). Los anticuerpos dirigidos contra esta proteína han demostrado inhibir el crecimiento de tumores xenógrafos y de las células de cáncer de mama transformadas que sobreexpresan Her-2. Específicamente los anticuerpos murinos (MAB) 4D5 dirigidos contra el dominio extracelular son un potente inhibidor de células de cáncer de mama humanas que sobreexpresan Her-2. El anticuerpo murino ha sido humanizado insertando regiones de complementariedad en una IgG, con la idea de hacerlo menos inmunogénico. El nombre de este anticuerpo Mab humanizado es trastuzumab y ha sido comercializado para su utilización en los casos de cáncer de mama que sobreexpresan dicho receptor. Una sobreexpresión de la proteina HER-2 ha sido señalada en aproximadamente el 30% de los cánceres de mama invasivos, y de ellos el 90% presentaban amplificación del gen. La función exacta de la porción extracelular del receptor no es bien conocida, pero se especula con su papel como estimulante de la proliferación celular o de los mecanismos de motilidad. Además, diversos estudios han demostrado el papel pronóstico de la expresión del gen Her-2, de modo que su sobreexpresión se asocia con un peor pronóstico y se correlaciona con otros factores pronóstico adversos como la negatividad de los receptores de estrógeno, la alta fracción de la fase S, ganglios axilares positivos, mutaciones de p53 y grado nuclear alto. En resumen que la sobreexpresión de Her-2 se presenta como un factor pronóstico de mayor importancia en la decisión del tratamiento adyuvante, y que la utilización terapéutica del Herceptin tiene una indicación formal en la enfermedad avanzada en pacientes que sobreexpresen Her-2, teniendo su máxima actividad cuando se combina con taxanos, si bien el regimen idóneo está por establecer. Para un futuro será además prioritario establecer su eficacia en los tratamientos de adyuvancia.
Otro anticuerpo monoclonal es el Cetuximab ó IMC-225 dirigido frente al EGFR. Este anticuerpo monoclonal ha mostrado en tumores xenógrafos humanos de células cancerosas epidermoides sinergismo con la radioterapia y la quimioterapia y su desarrollo en el ser humano se ha dirigido fundamentalmente al cáncer de cabeza y cuello y al cáncer colorrectal. Los estudios de fase I han mostrado su extraordinaria tolerancia, lo que ha dado lugar a estudios de fase II y III. Concretamente, en el cáncer colorrectal refractario a Irinotecán, Cetuximab ha mostrado actividad (17% de respuestas, y 31% de estabilizaciones) con mínima toxicidad (reacciones alérgicas G4 1%, y acné G3 en el 8%. Uno de los datos más importantes del Cetuximab, a diferencia del Trastuzumab, es que la respuesta obtenida es independiente del nivel de expresión del receptor, lo que sin duda establece un margen terapéutico mayor beneficiando a más pacientes. Actualmente se están llevando a cabo estudios de fase III que delimitarán su verdadera eficacia. Sin duda en el futuro se hace necesario establecer posibles marcadores biológicos que establezcan que el efecto biológico deseado se está consiguiendo. La tabla I muestra los posibles marcadores biológicos y el proceso celular en el que intervienen.
|
Tabla I.Posibles marcadores biológicos que pueden al menos teóricamente ser utilizados como efecto de la prueba biológica de fármacos con diana EGFR. |

En cuanto a los inhibidores específicos de la tirosina quinasa, los hay de tipo reversibles (quinazolinas) como el ZD1839 (Zeneca) y el OSI 358,774 (Roche) e irreversibles como el CI-1033 (pan-erb inhibitor) y el EKB 569 (erb1 y erb2). Las quinazolinas son de administración oral y ejercen una inhibición selectiva de los receptores del EGFR a nivel de la parte catalítica intracitoplasmática. Todo lo anterior produce una acumulación de p27/Kip1 y de la proteína Rb hipofosforilada con la consiguiente parada del ciclo celular en G1. El Iressa (ZD1839) es sin duda el inhibidor más desarrollado, siendo su toxicidad limitada (rash cutáneo, y escasas diarreas) y presentando una actividad prometedora en el cáncer de pulmón, tumor en el que ha sido más investigado.
El desarrollo del fármaco STI571 (Glivec-Imanitib) es el paradigma para el desarrollo clínico de fármacos basados en las nuevas dianas moleculares. STI571 fue diseñado como un inhibidor del receptor de crecimiento derivado de plaquetas PDGF-R, y pronto se observó que además es un potente inhibidor de todas las quinasas ABL, incluidas p210Bcr-Abl, p185Bcr-Abl, c-Abl e incluso la tirosina quinasa c-kit. Los primeros resultados han sido demostrados en leucemia mieloide crónica (LMC). El motivo es que en LMC existe una translocación del gen ABL (9q34) del cromosoma 9 al cromosoma 22, y también del gen BCR (22q11) del cromosoma 22 al 9. Esta translación recíproca determina la fusión de BCR y ABL y da lugar a una proteína tirosina quinasa quimérica P210 cuya función es inhibida por el STI571. Los resultados en la LMC han sido espectaculares, de modo que al fallo al interferón–alfa se produce el 96% de respuestas completas, de las cuales el 33% son de tipo citogenético. Incluso en las crisis blásticas es posible obtener un 39% de respuestas completas. Obviamente se precisa tiempo para conocer la verdadera dimensión de estas respuestas en lo que se refiere a la duración. Pero además de la LMC, el STI571 puede tener interés en muchos otros tumores que sobreexpresen PDGF o c-kit, como el cáncer de pulmón microcítico, el cáncer de próstata, sarcomas, gliomas, neuroblastomas, y tumores de células germinales. Concretamente los resultados presentados en la última reunión de ASCO en San Francisco en la primavera de 2001 son altamente interesantes. Los pacientes con tumores estromales gastrointestinales que expresaban el oncogén c-kit en el 100% fueron sometidos en dos estudios (uno denominado internacional y otro de la EORTC) al tratamiento con STI571. En el estudio internacional se encontró una respuesta parcial en el 54% (19/34), con una estabilización en el 34% (12/34); y en el estudio de la EORTC 26% de RP (4/15), y estabilización en el 53% (8/15). En suma que STI571 ha puesto de manifiesto que el desarrollo de fármacos a través de las nuevas dianas moleculares no sólo es posible sino efectivo, y abre la puerta a otras muchas posibilidades.
Los genes ras (H,K,N) están mutados en nada menos que el 30% de los tumores humanos y actúan como puntos clave en la transmisión de señalización intracelular como verdaderos interruptores. Así se pasa de una forma inactiva (off) en el que tiene un GDP fijado a otra activa (on) que tiene GTP. El proceso de activación está regulado por los factores GEFs (GDP/GTP exchange factors) y el de inactivación por los GAP (GTPase activating protein). En muchos modelos experimentales se produce una eliminación de la actividad GTPásica, y así permanece continuamente activa la proteína Ras. Un hecho fundamental es que Ras al igual que otras proteínas debe isoprenilarse para anclarse a la membrana, lo que se lleva a cabo a través de ciertas enzimas que le añaden un grupo farnesilo o un grupo geranilo-geranilo. La farnesilación corre a cargo de la enzima farnesil-transferasa, por lo que su inhibición podría acarrear la inactivación de Ras (Figura 6). Para ello se están utilizando péptidos miméticos que tanto en el oncorratón como en tumores humanos transplantados han mostrado una gran eficacia.
|
Figura 6. Los fármacos utilizados para bloquear la vía RAS actúan principalmente inhibiendo la farnesil-transferasa, enzima que a través de un fenómeno de isoprenilización ancla a la proteína RAS a la membrana, lo que es necesario para transmitir la señalización intracelular. |

Entre los inhibidores de la farnesil transferasa en fase de desarrollo clínico destacan los fármacos de Janssen-Cilag (R115777), el de Bristol-Myers Squibb (BMS214662) y el de Schering-Plough (SCH 66336). En el momento actual se están llevando a cabo estudios de fase I, fase II e incluso fase III. Concretamente, con el R115777 existe un estudio aleatorizado en pacientes con cáncer colorrectal avanzado que tras una o dos líneas de tratamiento quimioterápico los pacientes son aleatorizados para recibir dicho fármaco o tan sólo tratamiento de soporte. En el estudio se han incluido 345 pacientes y los resultados están pendientes. El objetivo primario es la supervivencia global y los secundarios la calidad de vida, el tiempo a la progresión y la respuesta objetiva.
QUINASAS DEPENDIENTES DE CICLINAS
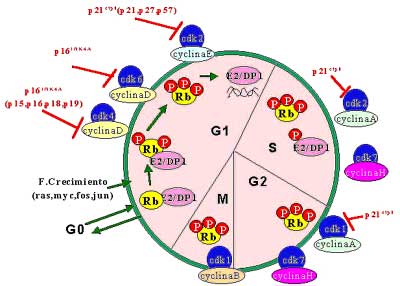
En la biología moderna es posible decir que el cáncer es la consecuencia de alteraciones en los procesos de señalización que en definitiva generan el control del crecimiento celular. En este aspecto es de suma importancia el ciclo celular, proceso que permite finalmente a la célula dividirse y donde intervienen decisivamente tanto señales de actividad positiva como son las quinasas dependientes de ciclinas (CDKs) y sus subunidades reguladoras , las ciclinas, como proteínas inhibidoras de las anteriores CDKs (familias INK y CIP7KIP) (Figura 7 y Figura 8).
|
Figura 7.La entrada en ciclo celular es compleja y se inicia con la fosforilización de la proteína retinoblastoma lo que permite liberar el factor de transcripción E2/DP1. Este fenómeno se encuentra catalizado por la acción de la ciclina D que a su vez necesita acoplarse a la quinasa dependiente de ciclina CDK4/6. Este complejo ciclina D/CDK4/6, se encuentra regulado por genes supresores del grupo p16INK4A y p21cip1. Posteriormente entran en acción en el resto del ciclo celular la ciclina E, la ciclina A y la ciclina B. |

Dentro del diseño de fármacos que actúan sobre el ciclo celular se han dividido en dos grandes grupos: 1) moduladores directos que inhiben la función catalítica de las CDK adecuadamente activadas, y 2) moduladores indirectos que alteran el estado activado de las CDK. Entre los moduladores directos se incluyen los siguientes subgrupos: 1.- Purinas y análogos de las purinas (6 dimetilaminopurina, isopenteniladenina, olomucina y roscoviotina), 2.- Productos sintéticos naturales (butirolactona, flavopiridol, saturospina, UCN-10, 9 hidroxi-elipticina, toyocamicina y suramina), 3.- Productos miméticos (p16, p21). A su vez entre los moduladores indirectos cabe destacar los siguientes: 1.- Los que actúan reduciendo los niveles de ciclina D (rapamicina, ansamicinas benzoquinóides, tirfostinas, inhibidores metabólicos), 2.- los que aumentan los inhibidores endógenos de CDK (butiratos, retinóides, y otros agentes diferenciadores), y 3.- los que alteran los puntos de control (checkpoints): inhibidores de fosfatasas (ac.okadáico), UCN-01 y la misma cafeina. Muchos de ellos se encuentran en fase I y fase II, siendo muy pocos los que están evaluándose en fase III.
|
Figura 8. Puntos críticos en la entrada en el ciclo celular (numerados de 1 a 7) donde al menos en teoría se pueden desarrollar agentes con misiones enormemente específicas. |

ONCOGEN SUPRESOR DE TUMORES p53
P53 es el guardián del genoma celular, y como gen supresor controla la división celular. Cuando p53 está activo detecta la aparición de daño celular y para la división de ésta para que se lleven a cabo los reglajes necesarios. De ser los daños sumamente intensos la célula gobernada por p53 entra en apoptosis. Ya se comprende que la pérdida de su función acarrea errores demasiado grandes y por tanto está directamente involucrado en el desarrollo de los tumores.
La introducción de p53 en las células con mutación del gen, mediante procedimientos de terapia génica es una posibilidad que está siendo analizada. Por el momento los estudios se limitan a tumores con crecimiento locorregional, y la única conclusión válida es que se trata de una excelente vía de experimentación, sin que los estudios fase I y II hayan mostrado una mayor relevancia.
Una de las áreas más excitantes de desarrollo terapéutico se refiere a los inhibidores de la angiogénesis tumoral así como inhibidores de varios receptores de factores de crecimiento. En la angiogénesis maligna existen varios componentes que incluyen la destrucción local, retracción de células endoteliales, mitosis de células endoteliales, migración de células endoteliales, interacciones matriz-células, estructura tridimensional, etc. Esta angiogénesis está balanceada entre los denominados factores de crecimiento endoteliales y los inhibidores endógenos de la angiogénesis. Los factores de crecimiento endoteliales son: 1) factor de crecimiento vascular endotelial (VEGF); 2) receptor VEGF-1 (flt-1); 3) receptor VGEF-2 (KDR), y 4) angiopoyetinas: angiopoyetina-1, receptor angiopoyetina-1. En cuanto a los factores inhibidores endógenos de la angiogénesis los más destacados son: 1) Trombospondina-1; 2) angiostatina; 3) endostatina: 4) vasostatina, y 5) interferón alfa2a y 2b.
Los principios básicos de este enfoque terapéutico son que los tumores mayores de 2 mm precisan la formación de nuevos vasos, que las células endoteliales de los vasos tumorales son diferentes de los vasos normales, y que los fármacos antiangiogénicos inducen la aparición de células dormidas y disminuyen el tamaño de los tumores.
El desarrollo de estos fármacos no ha hecho sino empezar. Si bien los estudios realizados en el animal son altamente esperanzadores, habrá que esperar a los resultados de los ensayos clínicos para conocer su verdadera eficacia y su mejor indicación que quizás podría ser en el tratamiento adyuvante y no tanto en la enfermedad avanzada.
Los fármacos antiangiogénicos pueden ser de varios tipos: 1) fármacos frente a los factores angiogénicos; 2) fármacos inductores de inhibidores endógenos de la angiogénesis (IEA), y 3) terapia génica con los IEA. En la angiogénesis tumoral hay varios pasos decisivos que pueden ser utilizados para el desarrollo de fármacos antineoplásicos. Los factores de crecimiento de tipo angiogénico, como el factor fibroblástico (FGF), el de transformación (TGF-a) y el vascular endotelial (VEGF), actúan como ligandos de los receptores de estos factores de crecimiento lo que da lugar a fosforilización de los residuos intracitoplasmáticos de tirosina-quinasa (actividad catalítica) iniciándose el proceso de señalización intracelular. Los receptores deben dimerizarse para su activación, y un hecho importante es que en los tumores existe sobreexpresión y transactivación de ellos. Finalmente la angiogénesis requiere la rotura de la matriz extracelular por colagenasas, fibronectina, laminina y glicoproteinas de membrana. La participación de las metaloproteinasas (constituida por enzimas degradativas) es esencial y en su producción colaboran estrechamente con las células tumorales, células estromales y endoteliales.
De los más de 20 fármacos antiangiogénicos actualmente en estudio, dos merecen la pena ser destacados por encontrase en fases más avanzadas de desarrollo. El primero es un anticuerpo monoclonal humanizado dirigido frente al VEGF. Terminada la fase I actualmente se ha completado la fase II correspondiente a un estudio aleatorizado comparando 5-FU/LV con 5-FU/LV más el anti-VEGF, estando pendientes los resultados. En cuanto a los inhibidores de tirosina-quinasa, el más sobresaliente es el SU5416 que al bloquear la fosforilización de la parte catalítica del receptor bloquea la señal de trasducción. Terminada la fase I y II están planificados los ensayos fase III en combinación con Irinotecán.
Por último es preciso decir que además de los fármacos diseñados a propósito con el fin antiangiogénico, existen otros que pueden también tener esta propiedad como son los inhibidores de señales de transducción (Ras) y las propias hormonas.
En definitiva, la biología molecular ha abierto unas puertas impensables hasta hace bien poco, que permitirán en primer lugar un mejor conocimiento del proceso tumoral, en segundo lugar llevar a cabo análisis de mutaciones hereditarias que impliquen el oportuno consejo genético, tercero un diagnóstico más ajustado desde un punto de vista patológico y molecular que nos ayudará a conocer con más detalles los factores pronóstico de la enfermedad y los factores predictivos individuales del paciente, y por último un enorme desarrollo terapéutico con aproximaciones a fármacos con mecanismo de acción y objetivos diferentes que obligan a un desarrollo clínico singular. Por lo tanto en lo que aquí concierne a esta revisión, el desarrollo de nuevos fármacos y la aplicación individualizada a cada paciente son los retos establecidos. El camino está abierto a todas estas posibilidades y es hora de ponerse en marcha.
1.- ASCO: Educational Book 2001. New agentes with new mechanism of action. 419-440, 2001.
2.- Baselga J.- The EGFR as a target for anticancer therapy-focus on cetuximab. Eur J Cancer suppl 4: S16-22, 2001. Review.
3.- Baselga J.- Targeting the epidermal growth factor receptor: a clinical reality. J Clin Oncol 19 (18 suppl) 41S-44S, 2001.
4.- Carter SK.- Clinical strategy for the development of angiogenesis inhibitors. The Oncologist 5 (suppl 1): 51-54, 2000.
5.- Denis LJ, Verwij J.- Matrix metalloproteinase inhibitors: present achievements and future prospects. Invest New Drugs 15: 175-185, 1997.
6.- Diasio RB, Johnson MR.- The role of pharmacogenetics and pharmacogenomics in cancer chemotherapy with 5-fluorouracil. Pharmacology 61: 199-203, 2000.
7.- Eisenhauer RA.- Phase I and II trials of novel anti-cancer agents: Endpoints, efficacy and existentialism. Ann Oncol 9: 1047-1052, 1998.
8.- Friend SH, Oliff A.- Emerging uses for genomic information in drug discovery. N Engl J Med 338: 125-126, 1998.
9.- Gelmon KA, Eisenhauer EA, Harris AL et al.- Anticancer agents targeting signalling molecules and cancer cell environment: challenges for drug development?. J Natl Cancer Inst 91: 1281-1287, 1999.
10.- Gibbs JB.- Mechanism based targer identification and drug discovery in cancer research. Science 287: 1969-1972, 2000.
11.- Hwang J, Sinicrope F, Safran H et al.- A phase II trial of Irinotecan and Trastuzumab (Herceptin) in patients overexpressing HER-2/neu in metastatic colorectal cancer. Pro ASCO 565, 2001.
12.- Korn EL, Arbuck SG, Pluda JM et al.- Clinical trial designs for cytostatic agents: Are new approaches hended?. J Clin Oncol 19: 265-272, 2001.
13.- Meropol NJ.- Novel targets in colorectal cancer. Educational Book. ASCO 2000. pp: 631-638.
14.- Miller LL, Elfring GL, Hannah AL et al.- Efficacy results of a phase I/II study of SU5416, 5-Fluorouracil (F) /leucovorin (L) relative to results in random subsets of similar patients from a phase III study of Irinotecan F/L or F/L alone in the therapy of previously untreated metastatic colorectal cancer. Pro ASCO 571, 2001.
15.- Senderowicz AM, Sausville EA.- Preclinical and clinical development of cyclin-dependent kinase modulators. J Nat Cancer Inst 92: 376-387, 2000.
16.-Mendelsohn J, Baselga J.- The EGFR receptor family as targets for cancer therapy. Oncogene 19: 6550-65, 2001.
17.- Shawver LK.- Tyrosine kinase inhibitors: from the emergence of targets to their clinical development. ASCO Educational Book, 29-44, 1999.
18.- Saltz L, Rubin M, Hochster H et al.- Cetuximab (IMC-225) plus Irinotecan (CPT-11) is active in CPT-11 refractory colorectal cancer that expresses epidermal growth factor receptor (EGFR).- Pro ASCO 7, 2001.
19.- Stoehlmacher J, Brabender J, Shirota Y et al.- Association between mRNA expression level of epidermal growth factor receptor (EGFR), immunohistochemistry and response to the EGFR-inhibitor C225 in advanced colorectal carcinoma. Pro ASCO 593, 2001.
20.-Von Hoff DD.- New agents with new mechanism of action and clinical activity. ASCO Fall Educational Conference Book, pp 122-126, 1999.
21.- Workman P.- Towards genomic cancer pharmacology: Innovative drugs for the new millennium. Current Opinion in Oncologic, Endocrine and Metabolic Investigational Drugs 2: 21-25, 2000.
Entry filed under: Cáncer en General, Tratamientos.

{kind=link}
Trackback this post | Subscribe to the comments via RSS Feed